Patient case #9 - Sickle cell crisis
Description
A young Nigerian woman came to the emergency room with intense pain in her limbs, chest, and stomach. She experienced dizziness, shortness of breath and headache. She was a known sickle cell disease patient and was immediately hospitalized and blood samples were drawn.
CBC results:
| Test | Results | Unit |
|---|---|---|
| WBC | 28 | x109/L |
| Hemoglobin | 87 | g/L |
| MCV | 105 | fL |
| PLT | 123 | x109/L |
A blood smear was made and scanned on a CellaVision® DC-1 analyzer.
Smear analysis on CellaVision® DC-1:
| WBC differential | % | x109/L |
|---|---|---|
| Neutrophils | 59,1 | 16,5 |
| Eosinophils | 0,9 | 0,3 |
| Lymphocytes | 22,7 | 6,4 |
| Monocytes | 17,3 | 4,8 |
| NRBC | 12/100 WBC | |
In the CellaVision® Remote Review Software, the Med-Tech found somewhat normal neutrophils, slightly reactive lymphocytes, normal monocytes and several nucleated red blood cells.
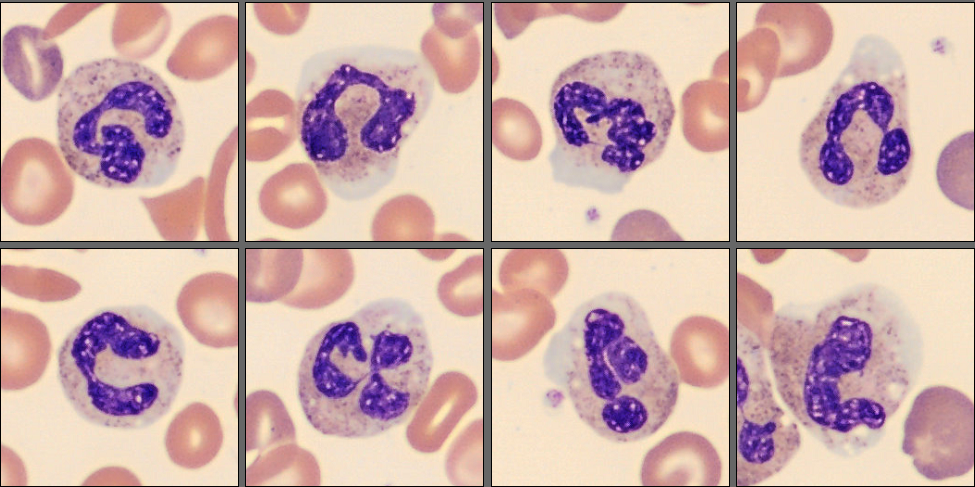
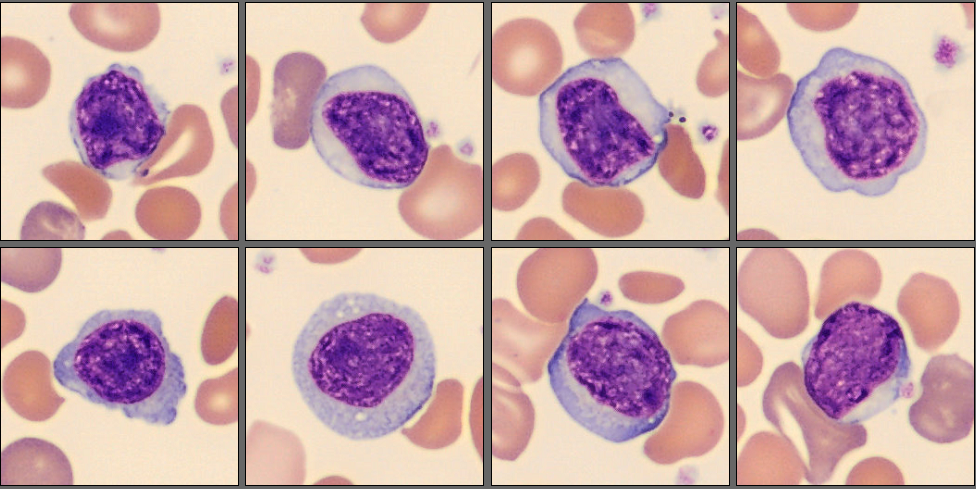
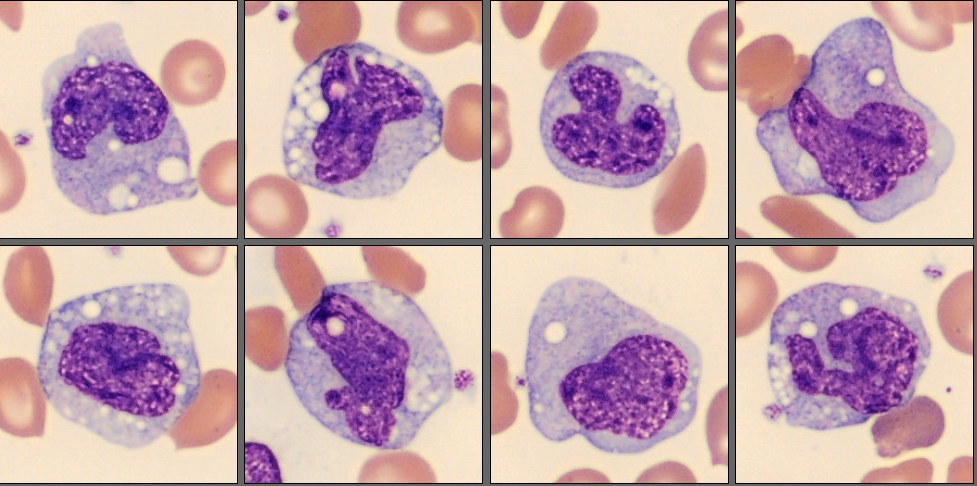
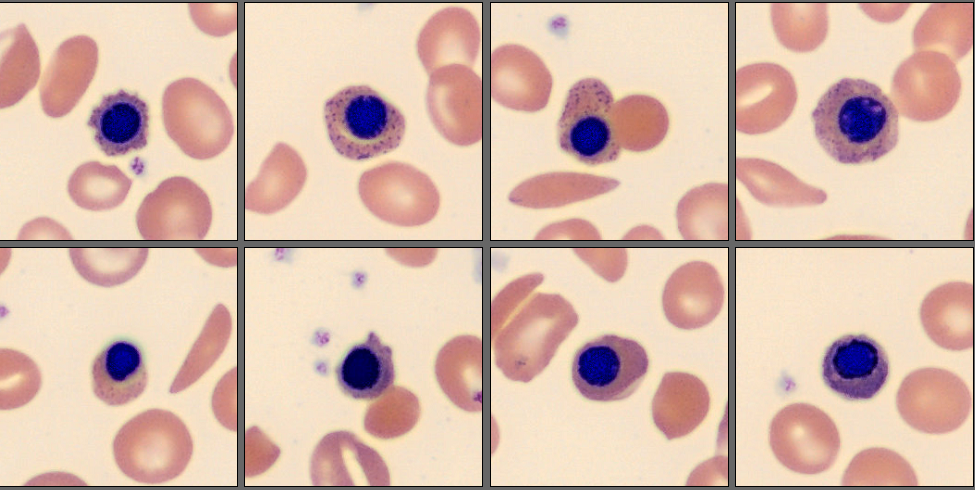
Unsurprisingly, in the RBC overview, they found a large population of sickle cells, some target cells and a larger population of polychromatic erythrocytes. In the RBC tab of the software polychromasia and anisocytosis was also flagged.
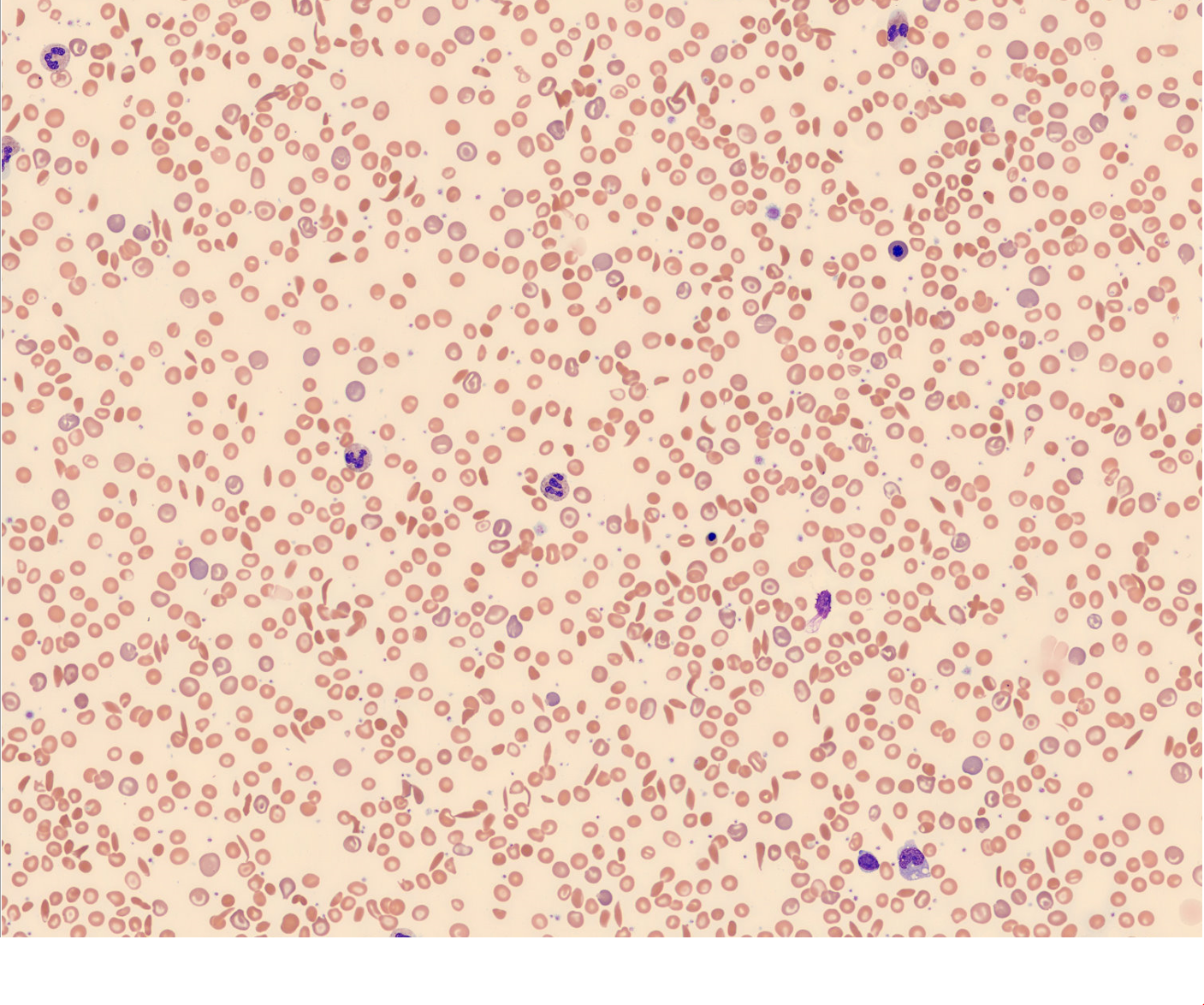
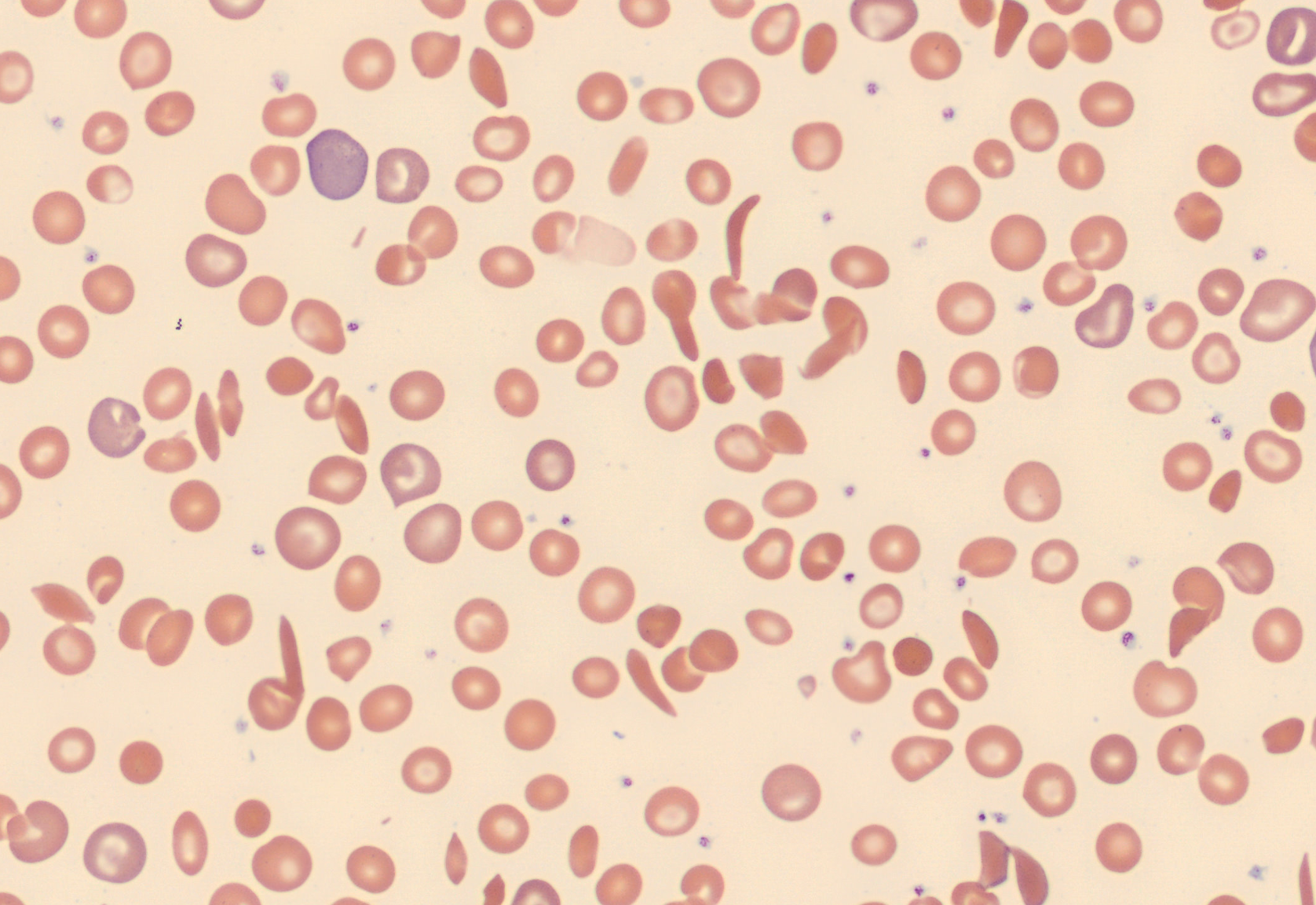
Diagnosis
Sickle cell crisis
Discussion
As she was a known Sickle cell patient, the emergency staff could immediately take care of the patient and give her pain relief and oxygen.
Sickle cells are elongated, sometimes crescent-shaped red cells with pointed ends. Boat-shaped cells have the appearance of a canoe viewed from above with sharpened ends. They lack central pallor, and stain deeply. Typically, 10-15 um in length with width narrower than normal cells. [1][2]
The sickling of the cells is caused by the hemoglobin S which is prone to polymerize in conditions of low oxygen.
The typical blood film shows anisocytosis, hypo- and polychromasia, sickle cells, boat cells, target cells, basophilic stippling, and NRBCs. Sometimes irregularly contracted cells and/or spherocytes are found. Many patients with sickle cell disease develop hypersplenism which shows after infancy, and this is evidenced by findings of Howell-Jolly bodies and Pappenheimer bodies. The WBC count is often slightly higher than normal range and occasionally it is possible to see phagocytosed RBCs in neutrophils and monocytes. During a crisis, the WBC count can rise sometimes up to 40-50 x 10^9, a neutrophilia, and an increase of NRBC and reticulocyte count. The affected red blood cells have a shorter life span of 10-20 days, (normal RBC have a life span of 90-120 days) which causes an acute worsening of the patient’s anemia [2]
Sickle cell disease affects more than 8 million people worldwide and is most common in people originating from Africa, Middle Eastern, Mediterranean, Central and South America and South Asia. [1]
Sickle cell disease is a group of inherited disorders caused by homozygosity for the β chain variant, hemoglobin S, or sickle cell hemoglobin. Normally the red blood cells are flexible and can easily move through even the thinnest blood vessels, but in sickle cell disease the affected red blood cells are misshaped to the crescent or sickle-shaped form which do not bend, and they can no longer move easily through the vessels and will block and/or slow down blood flow. Blockage of blood vessels means that the RBC will no longer deliver oxygen to the body, and this can cause severe pain which can start without warning, this is called sickle cell crisis. [1][2]
Early symptoms are jaundice (yellowish color of the skin) due to hemolysis, tiredness due to anemia and dactylitis (painful swelling of hands and feet) due to blocked blood vessels. The symptoms can worsen quickly, and they can experience severe pain, fatigue, shortness of breath, dizziness, fever, chest pain, coughing, and several other symptoms. [1]
There are several different ways to manage the sickling of the RBC. Hydroxyurea, which is an oral medication, is used to reduce sickling of the cells and can help prevent sickle cell crisis. Other oral medications are L-glutamine and crizanlizumab-TMCA. [1]
Another treatment is blood transfusion which is commonly used when patients have severe anemia and also for patients that have suffered from a stroke. [1]
Blood and bone marrow transplant is a potential curative therapy, but it is necessary to find a well-matched donor to be successful, and this is a high-risk procedure. [1]
Two types of gene therapy are now approved and aim to cure the patient from sickle cell disease. However, this is a costly treatment which requires weeks in hospital, and costly medications. These treatments are only available in a limited number of medical centers. [1]
___
Reference:
[1] NIH, Sickle cell disease (September 30, 2024) https://www.nhlbi.nih.gov/health/sickle-cell-disease# (Accessed 2025-05-14)
[2] Bain, Barbara J., (2015) Blood Cells, A Practical Guide (5th edition), John Wiley & Sons Ltd, p. 312-318; ISBN: 978-1-118-81733-9