Patient case #11 - Acute Promyelocytic Leukemia (APL)
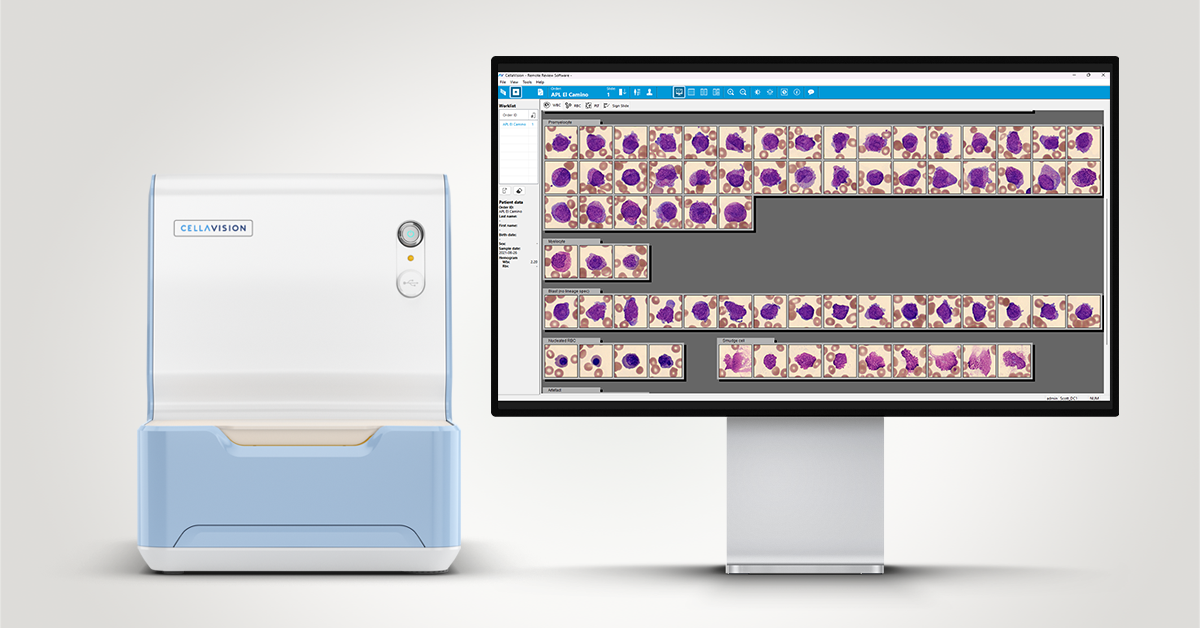
Description
A 55-year-old man came to the hospital with a rapid onset of fever, headache, nausea, vomiting, back pain, generalized weakness, and bleeding gums. His symptoms had developed over a short time frame, prompting urgent laboratory evaluation.
CBC results:
| Test | Results | Unit |
|---|---|---|
| WBC | 2,2 | x109/L |
| HGB | 77 | g/L |
| MCV | 82 | fL |
| PLT | 23 | x109/L |
A differential review on the CellaVision® DC-1 slide showed the following distribution:
| WBC differential | % | x109/L |
|---|---|---|
| Neutrophils | 8,8 | 0,2 |
| Lymphocytes | 40,7 | 0,9 |
| Promyelocytes | 33,6 | 0,7 |
| Myelocytes | 2,7 | 0,1 |
| Blasts | 14,2 | 0,3 |
| Non WBC | Count | Ratio |
|---|---|---|
| NRBC | 4 | 3,6 |
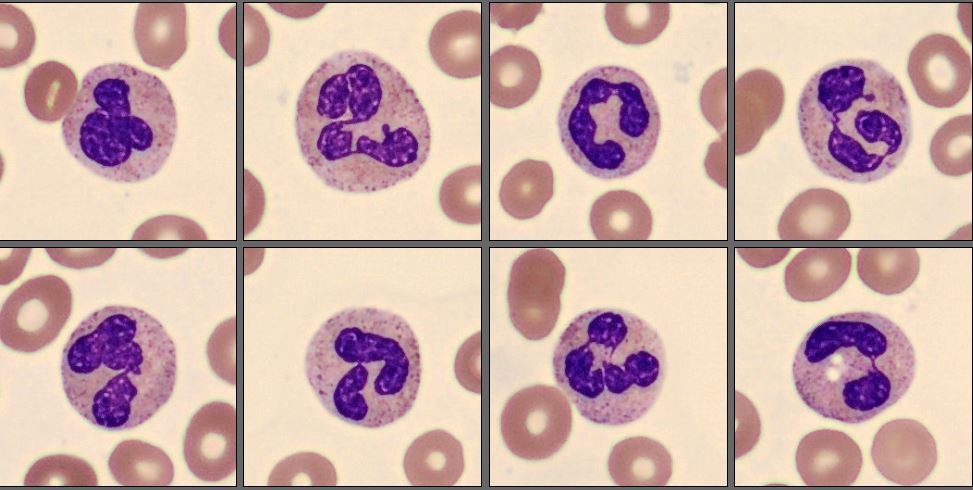
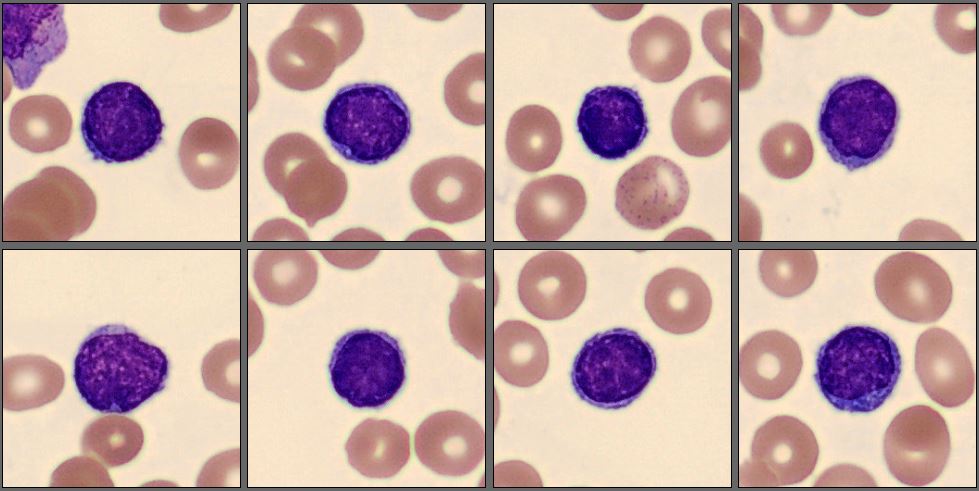
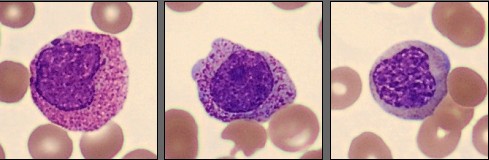
In this differential, only a few mature neutrophils and lymphocytes can be seen, while the smear is dominated by a high number of abnormal promyelocytes and blast cells. These immature cells are challenging to distinguish from each other, showing overlapping features and atypical morphology. In addition, myelocytes are present in very low numbers, indicating a disruption in normal myeloid maturation.
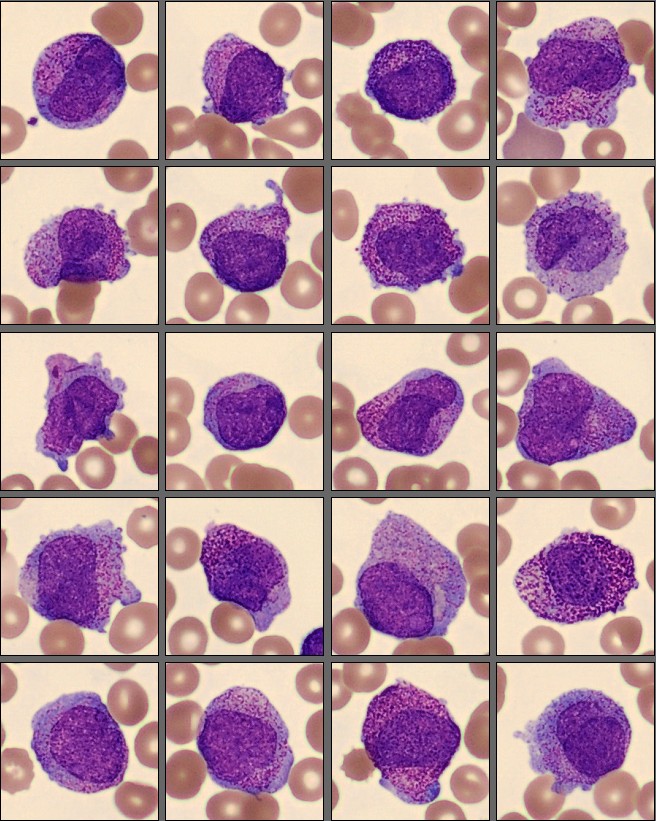

Molecular testing demonstrated a PML-RARA fusion.
Flow cytometry showed expression of CD13, CD33, and CD117, while CD34 and HLA-DR were negative. CD15, CD11a, CD11b, and CD11c were also negative.
Diagnosis
Acute Promyelocytic Leukemia (APL)
Discussion
Acute promyelocytic leukemia (APL) is a distinct subtype of acute myeloid leukemia (AML) characterized by the accumulation of abnormal promyelocytes due to the t(15;17)(q24;q21) translocation, resulting in the PML::RARA fusion [1]. Morphologically, APL is recognized by a predominance of atypical promyelocytes in the bone marrow and peripheral blood, although there is considerable variability between the hypergranular (typical) and microgranular (variant) forms [2].
In the hypergranular form, abnormal promyelocytes are large cells with abundant cytoplasm filled with coarse azurophilic granules, often containing multiple Auer rods that can form so-called faggot cells. Nuclei are usually irregular, folded, or bilobed, with fine chromatin and visible nucleoli. The microgranular variant, on the other hand, displays cells with scant or absent visible granulation and a high nuclear-to-cytoplasmic ratio, making them morphologically similar to blasts [3].
In this case, the peripheral smear demonstrated a predominance of abnormal promyelocytes and blast-like cells, with only a few mature neutrophils and lymphocytes. Many of the immature cells showed delicate chromatin, prominent nucleoli, and subtle cytoplasmic granulation, features that blur the distinction between promyelocytes and blasts. Myelocytes were scarce, suggesting a maturation arrest at the promyelocytic stage.
An important morphological consideration in APL is that the abnormal promyelocytes themselves are blast equivalents. In other words, the distinction between blasts and promyelocytes, which is diagnostically relevant in other AML subtypes, is less meaningful here because both represent the leukemic clone arrested at the promyelocytic stage [4]. Recognizing this concept helps explain why even experienced morphologists may find it difficult, and why molecular confirmation of PML::RARA fusion is essential to establish the diagnosis.
Immunophenotyping typically shows positivity for CD13, CD33, and CD117, with negativity for CD34 and HLA-DR, forming a characteristic APL profile [5]. The presence of PML::RARA fusion confirms the diagnosis and guides immediate initiation of targeted therapy with all-trans retinoic acid (ATRA) and arsenic trioxide (ATO). Early recognition is crucial, as APL is a medical emergency due to the high risk of disseminated intravascular coagulation (DIC), yet it is also one of the most curable forms of AML when treated promptly [6].
Morphology remains the first and most vital step in recognizing APL, and often the first opportunity to save a patient’s life. A skilled observer identifying abnormal promyelocytes on a peripheral smear can trigger urgent molecular testing and life-saving treatment within hours. In this context, digital morphology systems such as CellaVision® play a critical role in supporting the diagnosis of APL and many other types of leukemia.
The combination of expert morphologic insight and cutting-edge digital technology exemplifies how early laboratory detection can directly impact patient survival in acute leukemia.
Reference:
[1] WHO Classification of Tumours Editorial Board. WHO Classification of Tumours: Haematolymphoid Tumours. 5th ed. Lyon: International Agency for Research on Cancer; 2024.
[2] Bain BJ. Blood Cells: A Practical Guide. 5th ed. Wiley Blackwell; 2015.
[3] Arber DA, Orazi A, Hasserjian R, et al. The 5th Edition of the WHO Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36(7):1703–1719.
[4] Khoury JD, Solary E, Abla O, et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Blood. 2022;140(11):1200–1228.
[5] Falini B, Mecucci C, Tiacci E, et al. Acute Promyelocytic Leukemia: Diagnostic Immunophenotypic and Molecular Features. Haematologica. 2021;106(1):12–22.
[6] Sanz MA, Fenaux P, Tallman MS, et al. Management of Acute Promyelocytic Leukemia: Updated Recommendations from an Expert Panel of the European LeukemiaNet. Blood. 2019;133(15):1630–1643.