Patient case #3 - Thalassemia
Description
A 25-year-old Asian American female with a long history of anemia was seen in the emergency room with acute onset of fatigue, weakness, shortness of breath, jaundice, and hepatosplenomegaly. Her history includes recurring leg ulcers and gall stones. She is transfusion dependent.
Results:
CBC & Retic
| Test | Result | Units |
|---|---|---|
| WBC | 13.4 | x109/L |
| RBC | 2.25 | x1012/L |
| HGB | 5.7 | g/dL |
| HCT | 19.6 | % |
| MCV | 87.1 | fL |
| MCH | 25.3 | pg |
| MCHC | 29.0 | g/dL |
| PLT | 378 | x109/L |
| Retic % | 16.0 | % |
| Retic # | 360 | x109/L |
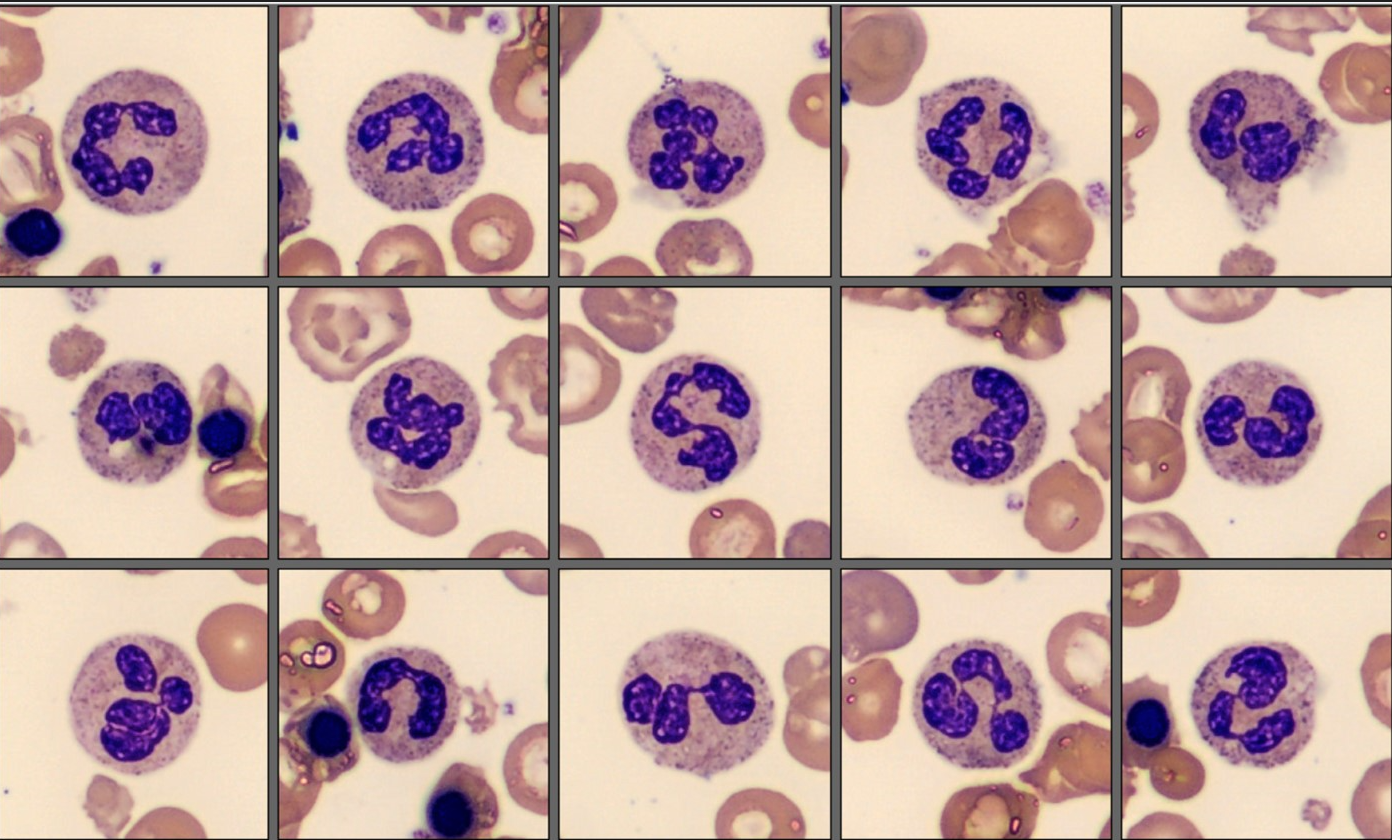
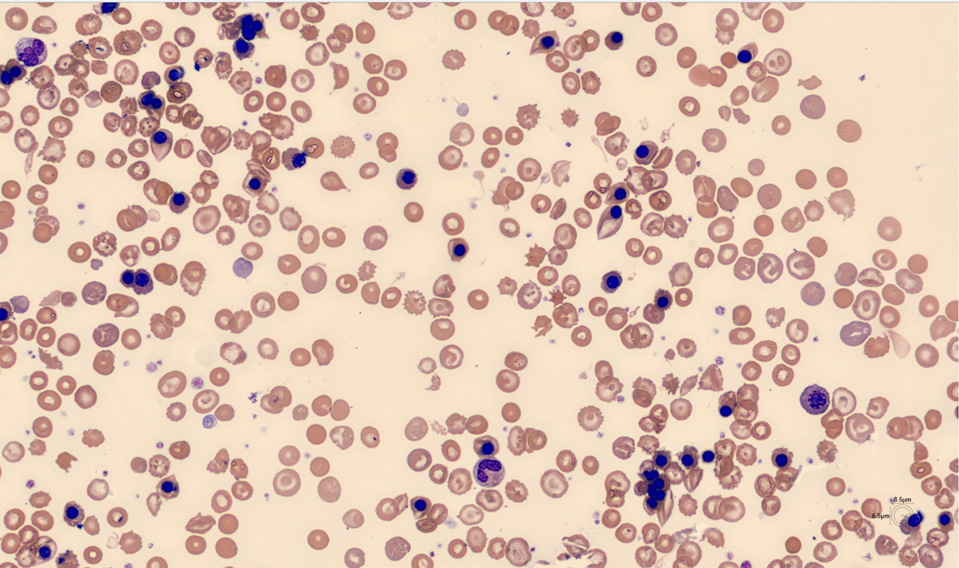
Left Shift and NRBC flags led to blood film analysis on Cellavision DC-1:
| Cell Class | % |
|---|---|
| Segmented neutrophils | 46.8 |
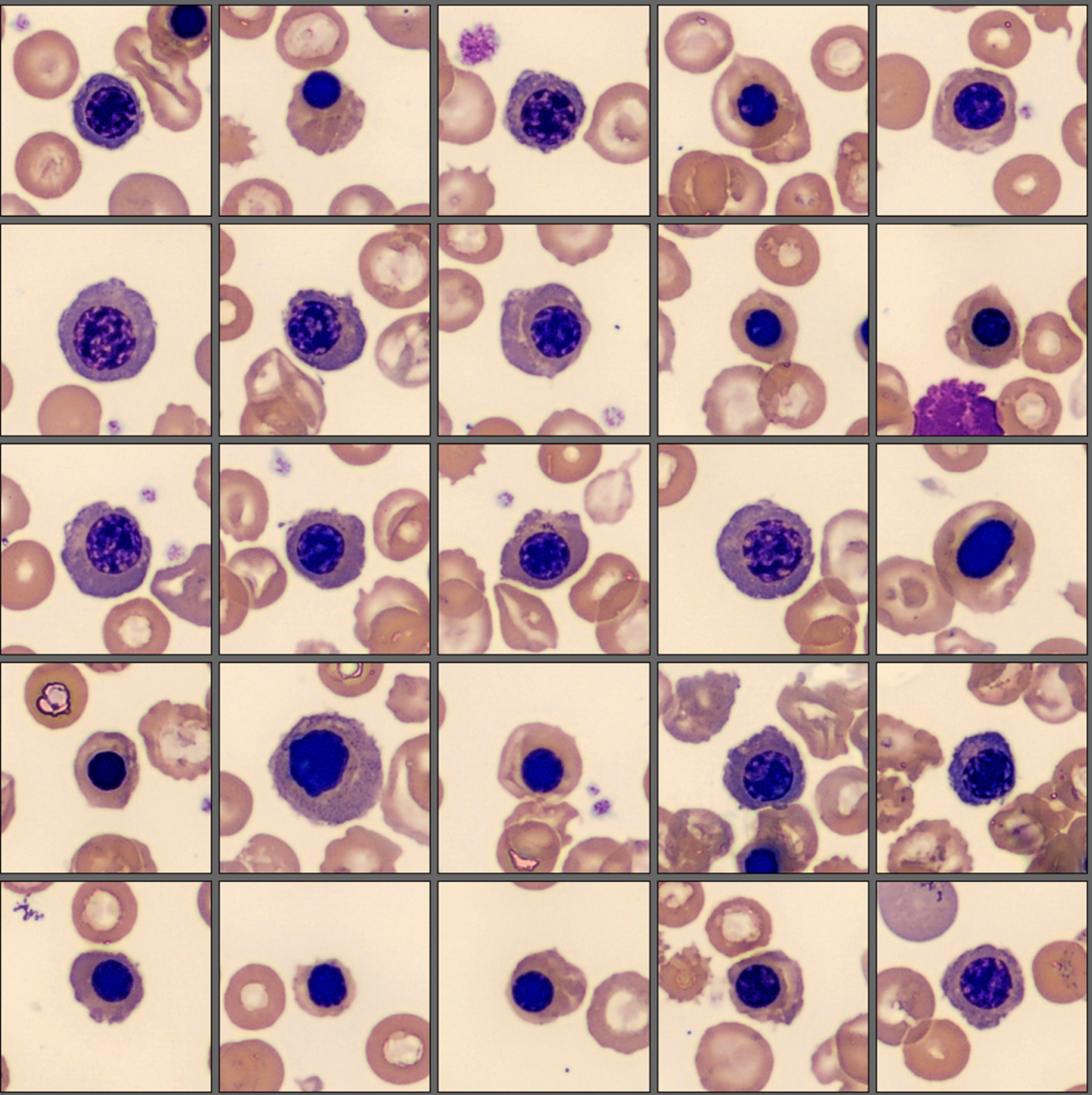
| Lymphocytes | 29.8 |
| Monocytes | 6.4 |
| Eosinophils | 4.3 |
| Basophils | 2.1 |
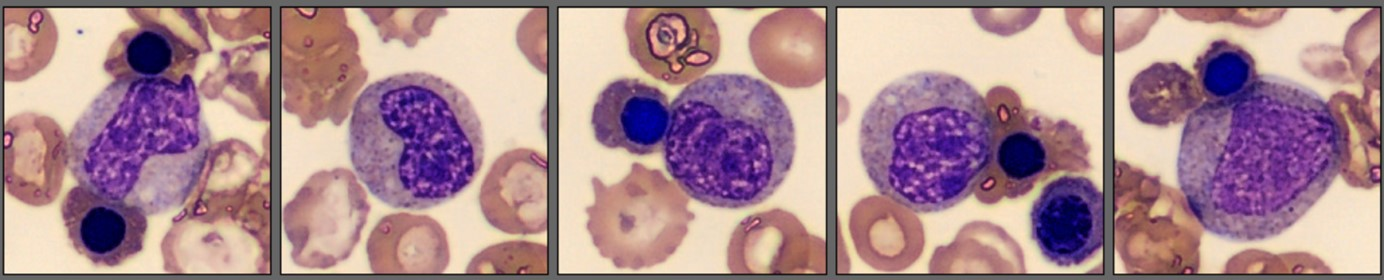
| Myelocytes | 4.3 |
| Metamyelocytes | 6.4 |
| NRBCs per 100 WBC | 1,830 |
Biochemical tests:
| Test | Result | Units |
|---|---|---|
| Bilirubin | 7.4 | mg/dL |
| LDH | 380 | U/L |
| AST | 49 | U/L |
| ALT | 65 | U/L |
| Haptoglobin | < 8 | mg/dL |
| Ferritin | 1655 | ug/L |
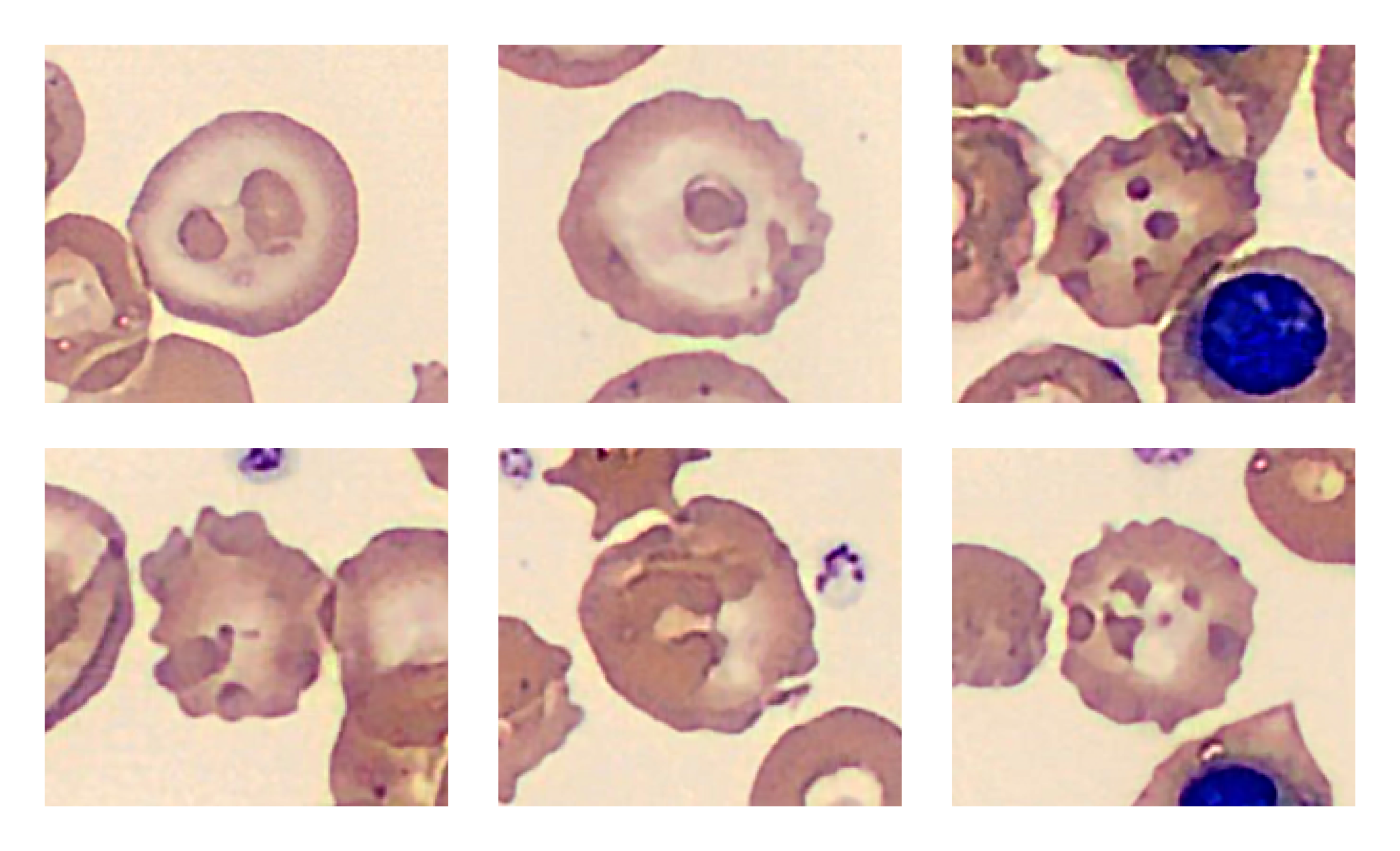
A blood film stained with brilliant cresyl blue revealed several RBCs with a “golf-ball” appearance characteristic of Hemoglobin H (Hb H).
HPLC: Sharp peak early in the F window (0.18 min) consistent with Hb H and small peak at 4.7 min suspicious for Hemoglobin Constant Spring (CS) . Molecular testing confirmed deletions in 2 alpha genes and CS mutation.
Diagnosis/Summary
The patient was subsequently diagnosed with alpha thalassemia: Hemoglobin H – Constant Spring disease (Hb H-CS) in hemolytic crisis.
Thalassemia is an inherited disease that results in a deficiency of globin chains, which are essential components of hemoglobin. Alpha thalassemia affects five percent of the world's population and 23 percent of people in Southeast Asia.1 Alpha thalassemia is associated with various degrees of anemia and a range of clinical symptoms depending on the genetic abnormalities present.
Hb H-CS is a form of alpha thalassemia in which two of four alpha globin genes are deleted and a third alpha gene is mutated. These lead to a reduction in normal alpha globin chains and the production of an elongated alpha globin chain called Constant Spring (CS), respectively.
In Hb H-CS, beta globin chains are produced at normal rates and because there are fewer alpha chains available to combine with, beta chains aggregate into unstable tetramers called hemoglobin H. These aggregates oxidize and damage RBC membranes which shortens RBC survival. The abnormal CS globin chains are also unstable and further damage cell membranes.2 The RBC deformities shown in the picture may be due to membrane-bound oxidized CS and beta globin chains.2
RBCs in Hb H-CS are overly hydrated and larger than typical thalassemic RBCs, which, together with the reticulocytosis, helps explain the normal MCV in this patient. In addition, the RBCs appear hypochromic because they contain significantly less hemoglobin than healthy cells. Insufficient production of normal hemoglobin and shortened red cell survival lead to lifelong anemia.
As can be seen in this case, patients with Hb H-CS disease experience episodes of hemolytic crisis, as evidenced by clinical symptoms, severe anemia, reticulocytosis, bilirubinemia and other biochemical indicators. A steep drop in Hb levels is often triggered by an infection and/or fever. The high ferritin level is associated with excess storage iron and is common in Hgb-H-CS patients who receive regular transfusions. These patients often require chronic iron chelation therapy.
___
1. Goh LPW, Chong ETJ, Lee PC. Prevalence of Alpha(alpha)-Thalassemia in Southeast Asia (2010-2020): A Meta-Analysis Involving 83,674 Subjects. Int J Environ Res Public Health. Oct 9 2020;17(20)
2. Schrier SL, Bunyaratvej A, Khuhapinant A, et al. The unusual pathobiology of hemoglobin constant spring red blood cells. Blood. Mar 1 1997;89(5):1762-9.